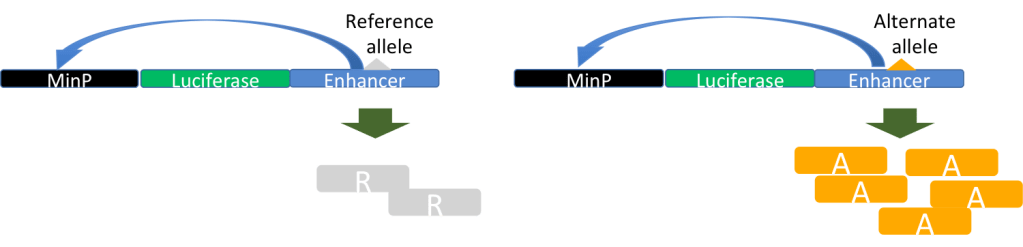
Identifying the phenotypic effect of nucleotide substitutions in non-coding regions is a recognized challenge in functional genomics and human genetics. Our contribution to these research questions is two-fold: one is participation in the development of statistical and computational methods to predict the function of non-coding variants, through collaborative projects with Xiaoquan Wen (University of Michigan) and Roger Pique-Regi (Wayne State University), and the other is experimental validation of the computational annotations of non-coding variants. Finally, once we have identified functional non-coding variants in the human genome, we use population genetics analyses to understand their evolutionary relevance (Wen et al, 2015, PLOS Genetics; Wen et al, 2016, AJHG; Moyerbrailean et al, 2016, PLOS Genetics; Findley et al, 2021, PLOS Genetics).We have developed experimental and computational methods for biallelic high-throughput reporter gene assays and electrophoretic mobility gel shift assays (Kalita et al, Bioinformatics, 2017; Kalita et al, 2018, Genome Res). These assays will allow us to validate also genetic variants that we and others have fine mapped in molecular and complex traits association studies, and to experimentally validate our novel statistical methods (Wen et al 2015, 2016, 2017). We are now extending these experimental approaches to investigate allele-specific effects on gene regulation in different environmental contexts, for human and Neanderthal sequences.
